I missed an important detail: """an academic team has developed its own protein-prediction tool inspired by AlphaFold 2, which is already gaining popularity with scientists. That system, called RoseTTaFold, performs nearly as well as AlphaFold 2, and is described in a paper in Science paper also published on 15 July"""
One of the things I say about CASP has to be updated. It used to be "2 years after Baker wins CASP, the other advanced teams have duplicated his methods and accuracy, and 4 years after, everything Baker did is now open source and trivially reproducible"
now, it's baker catching up to DeepMind and it took about a year
I see an interesting geometric similarity between the two models, namely the attention mechanism that learns relationships between structure in embedded reference frames (i.e. 1D,2D embeddings in RoseTTaFold or "local" frames in Alphafold) and the true structure of the intrinsic space reference frames (3D coordinates in RoseTTaFold and "global" frames in Alphafold)
Also announced today was RoseTTAFold from UW's Baker Lab, which claims nearly the same accuracy at much higher efficiencies. There's a public server and paper in Science.
> With RoseTTAFold, a protein structure can be computed in as little as ten minutes on a single gaming computer.
I guess its a little less accurate but the quick compute time makes as much difference too. E.g. research students can have multiple less costly mistakes before achieving what they want with the software.
A disturbing thing is that the architecture is much less novel than I originally thought it would be, so this shows perhaps one of the major difficulties was having the resources to try different things on a massive set of multiple alignments. This is something an industrial lab like DeepMind excels at. Whereas universities tend to suck at anything that requires a directed effort of more than a handful of people.
>A disturbing thing is that the architecture is much less novel than I originally thought it would be, so this shows perhaps one of the major difficulties was having the resources to try different things on a massive set of multiple alignments.
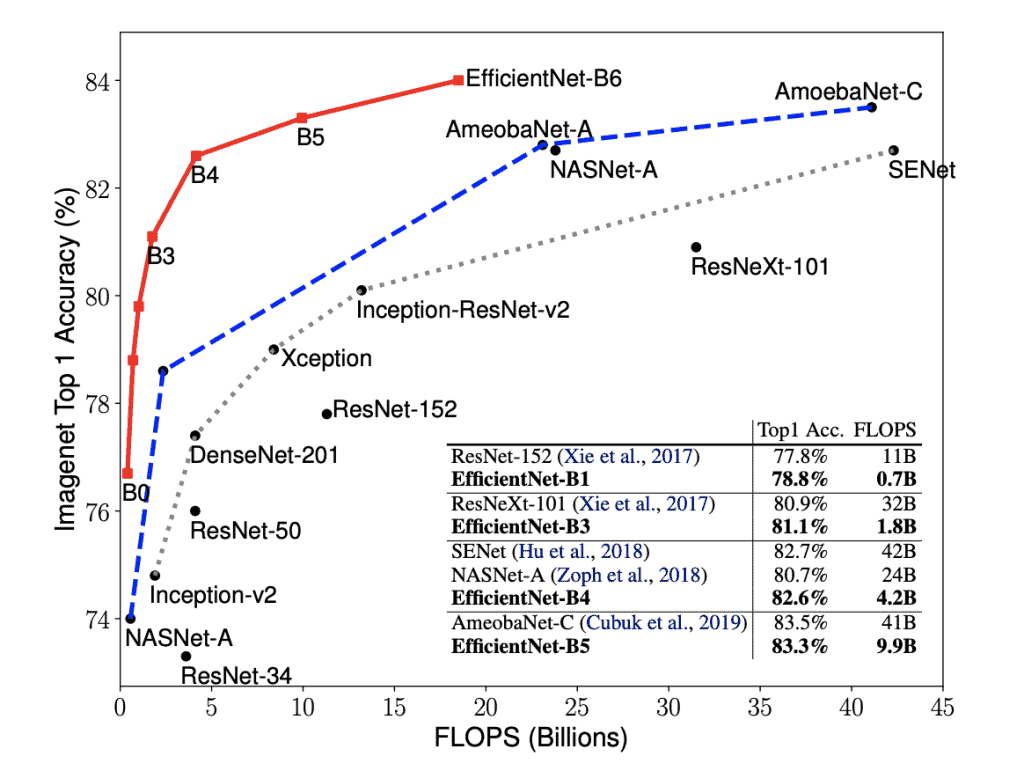
Most of the compute in ML research seems to be going into architecture search. Once the architecture is found, training and net finetuning/transfer learning is comparatively cheap, and then inference is cheaper still. This implies we could see 10-100x gains in AI algorithms using today's hardware, or sudden surprising appearance of AI dominance in an unexpected field. (Object grasping in unstructured environments? Art synthesis?) A task could go from totally impossible to trivial in a year. In retrospect, the EfficientNet scaling graph should have alarmed more people than it did: https://learnopencv.com/wp-content/uploads/2019/06/Efficient...
Waymo has been puttering along for years, not announcing much of interest. This may have caused some complacency about self-driving cars, which is a mistake. Algorithms only get better, while humans stay the same. Once Waymo can replace some human drivers some of the time, things will start changing very quickly.
Self-driving is a cursed problem, but the thorniest obstacles relate to optics and politics and not technology. AI can already replace some human drivers some of the time; but it doesn’t matter because each news story about a Tesla killing a passenger or driving into a parked fire truck sets back public acceptance of self-driving cars by years.
True, and Tesla is so far behind competitors when it comes to self driving that they knowingly push the envelope, because either they succeed, or they kill their customers but hurt all self-driving companies. So both scenarios work to their advantage and they just pay the minor fines and compensations to the people the kill in the process. And the Elon posts some Tweets blaming the people who got killed and his fandome happily cheers.
There is a reason Waymo is progressing at what looks like snail speed from outside observers, and that reason is that it’s the only ethical pace for development and testing of what could eventually become broadly available fully autonomous vehicles.
Teslas philosophy is that self driving is useless if it can't handle every road and route out there, while Waymo who already has a ride hailing service, relies on pre-mapped routes and databases.
Both have their advantages and disadvantages. Waymo requires huge databases and a constant network connection. But it's good enough to be used in real-life without a backup driver, in certain select locations that is. If there's a road construction somewhere, don't expect the Waymo car to handle it unless they update their database.
Tesla on the other hand attempts to solve the problem with minimal database use, where most of the info coming from road markings and traffic signs. A much harder problem.
Going slowly may mean fewer people die directly. But I think it’s useful to remember that every year we delay being able to replace human drivers means that roughly a million lose their lives worldwide in auto accidents, and many more are maimed, many permanently.
It’s obviously complex, though, bad PR likely delays things as well.
> Most of the compute in ML research seems to be going into architecture search.
No it's not. Only Google spends significant time with automatic architecture search, and many people think this is really to try to sell cloud capacity.
> Once the architecture is found, training and net finetuning/transfer learning is comparatively cheap
Training isn't cheap for significant problems.
Getting the data is very expensive, and compute is a significant expense for large datasets.
> This implies we could see 10-100x gains in AI algorithms using today's hardware
Actually, most of the time we see 10-100% (percent! not times) gains from architecture improvements, whether they be manual or automatic.
But that is very significant, because a 10% improvement can suddenly make something useful that wasn't before.
>No it's not. Only Google spends significant time with automatic architecture search, and many people think this is really to try to sell cloud capacity.
Maybe not automatic architecture search but a lot does go into testing different architectures and changes to them in a more manual manner. Though yes, those tests are run on a smaller scale so for those huge models, training will be a bigger portion compared to architecture search than for smaller models.
If you don't you might not realize they are comparing ResNet (designed for ultimate performance) vs EffcientNet to show how close it gets in accuracy in the FLOPs budget.
Note that the best accuracy for EffcientNet is roughly 1-2% better than ResNet/ResNeXT/SENet/etc but does have a much better FLOPs budget.
But these other architectures were never optimised for FLOPs. It wasn't event a consideration when designing them.
And EffcientNet is about a (manually designed) technique for scaling neural networks up in accuracy. Only EffcientNet-B0 is designed by AutoML, the others are scaled up. See the paper[1] for complete details.
Like-for-like should be against MobileNet etc. EfficentNet is better, but the comparison is more reasonable in general.
> A disturbing thing is that the architecture is much less novel than I originally thought it would be, so this shows perhaps one of the major difficulties was having the resources to try different things on a massive set of multiple alignments. This is something an industrial lab like DeepMind excels at. Whereas universities tend to suck at anything that requires a directed effort of more than a handful of people.
Yeah, the HN commentary on Alphafold has a high heat-to-light ratio. I'm eager to read the paper because the previous description of the method sounded remarkably similar to methods that have been around for ages, plus a few twists.
The devil is going to be in the details on this one.
It means that the conversation isn't producing much of value despite lots of activity, like an inefficient lightbulb that wastes energy by emitting heat instead of light.
Incandescent light bulbs are generally very inefficient in producing light, compared to LED for example. They produce a lot of heat and not much light for which they are made.
So in this context I suppose that gp implies that these threads don't provide much meaningful discussion but rather lots of hand waving.
It’s trying to say light is more valuable than heat, or some such folksy thing. I cook steak in the dark so I don’t find it to be a very insightful metaphor.
Transformers seem to be the successor to conv nets. But in my direct experience advocating for them, it's amazing how reluctant industry peeps were to trying them because they associated all the limitations of LSTM networks with them for a long time. YMMV, but that's how it went with me.
I even predicted DeepMind's CASP 14 network would be transformer-based back in 2018, but I couldn't have told you the details of that transformer, just that it was a no-brainer to move from fixed width convolutions to arbitrary width attention sums because sequence motifs and long-range interactions are of arbitrary width in the sequence.
All that seems to have changed with AlphaFold 2 because unlike GPT-XXX, this isn't a parlor trick with memorized text. This is actually useful and the FOSSing of the network will spawn all sorts of new applications of the approach.
So now I wonder what will replace Transformers because nothing lasts forever and there are a lot of smart people trying all sorts of new ideas.
The key difference seems to be using the multiple alignments and assumption about evolutionary conservation? Useful for genes conserved, but less useful for de-novo proteins (like COVID and cancer) I guess?
Dunno yet. MSAs were always a key input to Rosetta (previous best method). How they were used was very different.
Fundamentally, everything in this space (= non-physical methods) is about inferring structure from things that are closely related. And you can't solve the problem at all for non-trivial proteins using physics, so here we are.
I guess poster is referring to purely using physics to work out the structure - i.e. no more knowledge than how atoms/molecules move and the sequence. At the moment knowledge is gleaned from evolution by virtue of evolutionary conservancy.
Essentially yes. In theory, you could start from quantum mechanics and get the structure of any chemical matter. In practice, that's intractable. So there has been decades of work to make ever-simpler, less expensive approximations, and use these models of forces/energies to predict the structures of complex biomolecules.For example, google "molecular mechanics" or "molecular dynamics", and you'll find discussion o f things like the van der waals model of an atom, models of chemical bonds that resemble springs from physics 101, coulomb's law, and so on.
People take these simple models, chain them together in different ways, adjust the free parameters (i.e. weights) by fitting to known data, and thus create "force fields" that can be used to estimate the energies and/or forces on a molecule, given only the coordinates of the atoms. These methods work OK for some limited problems, but tend to fly off the rails as simulation times get longer, or if atomic systems have more electrostatics, motion, "weird" atoms (like heavy metals), etc.
This (Alphafold) kind of work is completely different, in that it starts from data, and uses the physical models only to do final refinements (if at all).
That's the case with basically everything DeepMind does. They have a very good PR department which hypes up everything they do while conveniently ignoring that basically nothing of any practical consequence has come of their endeavors. But I do think it's important that these companies exist now so we can see what not to try going forward.
Well, the CASP14 results do speak for themselves. Protein structure prediction is not necessarily of great meaning to drug discovery or biology, but they pretty much blew everyone else out of the water in a fair contest. For that reason, they deserve praise.
It's a little like making a robot that is very, very good at something pointless (say, using a yo-yo). Who knows where it might lead, but if they make the best damned yo-yo bot in the world, they deserve whatever praise they get from the yo-yo community.
Their PR strategy is to take problems people thought were impossible to solve in the next 10 years, and solve them (Go) or nearly solve them (StarCraft 2, protein solving)
No one thought that Go was impossible except people who weren't involved in engineering/software and shouldn't have been taken seriously in the first place.
Superhuman game AI has existed for decades. It's entirely unsurprising that one can play a strategy game, and no one thought it wasn't possible.
I'll send you $500 USD if AlphaFold derivatives lead to a single new therapy or drug that helps real patients in the next 5 years.
'In the next 10 years' is the key phrase here. Everyone watching Go (I looked at the problem 15-ish years ago) thought it required a massive advance to beat the best humans. DeepMind made that advance.
'bah, whatever, both Newton and Leibniz were hacks. Everyone knew calculus would have been invented anyway. It's basically a consequence of some stuff Archimedes did.'
DM didn't "solve" anything around proteins. They just made an improvement to existing homology modelling methods. If you look, the system is incredibly dependent on having large numbers of high quality sequence alignments to proteins with known structure and lots and lots of evolutionary data.
This was actually fairly obvious 20 years ago and it's been disappointing to me to see how long it took somebody to make this improvement, but really, it couldn't have been done without recent algorithmic improvements and huge amounts of CPU time.
Well here's one example: Deepmind made Wavenet, which turned text-to-speech on it's head. Variants of Wavenet underlie most or all of the talking machines (Google assistant, Alexa, etc.).
Well, also Alpha-Fold, and let's not forget beating the world champions at Go, which was a really major unsolved problem. I'll also contend that WaveNet was not 'maybe-slightly-better,' but actually a massive leap forward.
The phrase 'who pissed in your cheerios' comes to mind... Here's hoping that if you ever build something worthwhile it is received with a bit more compassion.
I haven't read the full paper but there is certainly some new/exciting developments i'm seeing just from scanning. The “Invariant Point Attention" which is described as a novel, geometry-aware and equivariant attention operation is pretty huge.
Something along these lines was speculated to be to be used by Fabian Fuchs [0] soon after the original CASP competition. Basically, it's a huge win for the geometric deep learning people, and indicates an exciting direction for mainstream academia to move in.
I'm genuinely curious: could the output of Alphafold be fed into a classical folding algorithm (as a starting point), or is the output of Alphafold too far down the wrong path, in these cases?
many of these resources are available, it's mostly that academic scientists don't have the time, money, or expertise to manage large datasets. However, the community has maintained high quality MSA database for decades and that's exactly the work that DM drafted off.
> academic scientists don't have the time, money, or expertise to manage large datasets
I may be cynical about general expertise, as a support person, but large datasets have long been stock in trade of areas I'm more or less familiar with, whether "large" is TBs or PBs like CERN experiments. (When I were a lad, it was what you could push past the tape interface in a few days -- data big in cubic feet...)
Tape is worthless except for archival purposes (and it's not particularly good). it should not be the constraint on the dataset (IE, any important dataset should already be in live serving with replication).
Very few players wrangle petabytes effectively. Many players have petabytes, but they're just piles of disorganized data that couldn't be used for training ML. Moving petabytes is still a huge pain and few folks have proficiency in giving ML algorithms high performance access to the data.
Fantastic, they released the dataset and code to train the model. Science will be able to proceed.
edit: not the code to train the model, just the code to run inference.
The underlying sequence datasets include PDB strucrures and sequences, and how those map to large collections of sequences with no known structure (no surprise). Each of those datasets represents decades of thousands of scientists work, along with programmers and admins who kept the databases running for decades with very little grant money (funding long-term databases is something NIH hated to do until recently).
> The total download size is around 428 GB and the total size when unzipped is 2.2 TB. Please make sure you have a large enough hard drive space, bandwidth and time to download.
> This was tested on Google Cloud with a machine using the nvidia-gpu-cloud-image with 12 vCPUs, 85 GB of RAM, a 100 GB boot disk, the databases on an additional 3 TB disk, and an A100 GPU.
This is amazingly detailed for a researcher who wants to follow in the track and also Apache licensed, which is one road-bump out of the way for a commercial enterprise, like an actual drug manufacturer who wants to burn some money trying this out.
edit: said the last part too fast, the code has a "the AlphaFold parameters are made available for non-commercial use only under the terms of the CC BY-NC 4.0 license"
Yes, all science should be communicated in the form of an academic paper wiht a supporting git repo and quickly downloadable dataset and a fast path to reproducing the work. That would be a huge change from the establishment.
It's quite unclear what value this will have to pharma; personally I doubt this has any direct applications (and I'm one of the few people in the world that can say that with deep authority).
Surely not all science. Just as well Dirac wasn't required to communicate that way the equation that fundamentally underlies the phenomenon discussed, and you couldn't put the unique facility my thesis work pioneered into git! I do highly approve of publishing software and data where possible, of course, since before Free Software needed to be coined, and it's much easier now.
I've been doing protein pharma research and the structure is only a first step, then years of figuring out the kinematics and dynamics of the protein, figuring out how it works, how all the natural ligands bind and affect the kinematics.. and only after all that you might conceivably start to engineer drug compounds (unless you bootstrap by a natural ligand to tweak "randomly", but then again, that's how pharma development traditionally works).
Still, even if structure determination is not on the "critical path", it IS a big barrier that has (started) to fall now.
Yes, I skimmed the paper already and it wasn't too surprising. There are details that will take some time to parse out to understand how important they are.
Personally, I've found over decades that academic papers like that are far less useful to me than a github project and downloadable data that I can inspect, run and modify on my own. Other folks I know could read that paper and write the code in a day, I always wish I could do that.
This isn't a criticism - I'm just curious to hear people's thoughts on this. When I look at this code, one of my initial reactions is that it does not seem to be very thoroughly tested. Sure, certain modules have been tested (e.g. `model.quat_affine`) but it's not clear how completely. Meanwhile, other modules, for example `model.folding`, have not been tested at all, despite containing large amounts of complex logic. That kind of code that works with arrays is very easy to get wrong and bugs are difficult to spot.
My experience working with code written by researchers is that it frequently contains a large number of bugs, which brings the whole project into question. I've also found that encouraging them to write tests greatly improves the situation. Additionally, when they get the hang of testing they often come to enjoy it, because it gives them a way to work on the code without running the entire pipeline (which is a very slow feedback loop). It also gives them confidence that a change hasn't lead to a subtle bug somewhere.
Again, I'm not criticising. I am aware that there are many ways to produce high quality software and Google/DeepMind have a good reputation for their standards around code review, testing etc. I am, however, interested to understand how the team that wrote this think about and ensure accuracy.
In general, I hope that testing and code review become a central part of the peer review process for this kind of work. Without it, I don't think we can trust results. We wouldn't accept mathematical proofs that contained errors, so why would we accept programs that are full of bugs?
My understanding is that it has been manually tested. I.e. it has produced correct results to previously intractable problems. I'm not sure how much automated testing would add at that point.
Unit testing usually isn't easily replaced by manual testing. If you have, for example, 3 units that can be in 2 different modes each, that's 2^3 different combinations, but only 2*3 unit modes. Testing the end result is more work than testing the units.
Discovery science is different from web software engineering.
Most discovery scientists use manual testing, not unit testing. Very few actually do integration tests or system tests (this is something I'm trying to change).
And, given the external results of the application, it's unclear to me how much additional value would come from a rigorous testing system.
Sure, I'm trying to take the idea of merging continuous integration with workflow/pipelines. It's all stuff that I learned at Google and is non-proprietary. The idea is have presubmit checks that invoke a full instance of a complex pipeline, but on canned (synthetic or pseudoanonymized or somehow not directly connected to the prod system) data, as an integration test. This catches many errors that would be hard to debug later in a prod workfflow.
In a sense, I see software testing/big web data and modern large scale data processing in science as a continuum and I want to bring the practices from the big web data and testing fields to bear on science pipelines.
Prior to this model, protein folding hadn't seen significant advancements in a decade or more. Worrying about the lack of tests in a first of its kind model is very much akin to complaining about the choice of font in a user manual for the world's first warp drive. I understand you're attempting to frame the problem in terms in things you know, but trying to weigh down pioneering research with professional development ceremony is very much counterproductive. The 'missing' ceremony would not have contributed to the strength of AlphaFold's result, the model's only purpose was to compete within the context of an existing validation framework.
Research code is highly volatile: the details and architecture changes a lot. It is much more important to invest the time into writing more experimental code and validate it with e2e functional tests that don't need to change, than to constantly having to rewrite the code and the tests.
> The AlphaFold parameters are made available for non-commercial use only, under the terms of the Creative Commons Attribution-NonCommercial 4.0 International (CC BY-NC 4.0) license. You can find details at: https://creativecommons.org/licenses/by-nc/4.0/legalcode
Does CC BY-NC actually do this? As far as I can tell it only really talks about sharing/reproducing, not using.
Or is the only thing prohibiting other commercial use the words "available for non-commercial use only"?
Artbreeder has some interesting prior art here: nVidia forbid commercial use of StyleGAN, but artbreeder disregarded it and happily sold all the breeding you wanted. No one seemed to care.
I suspect that the clause is there to prevent a startup launching on the basis of “see this trained model? Yeah, that’s literally our business model” though, which is a mildly amusing thought, wot wot.
So basically, a few tens of thousands, sure. A few million, big G might have a problem.
Still, the smart move would be to launch the business anyway, and gamble that you can work out a licensing deal.
If you took their parameters, then trained it for while on a different set of data, it would vary from the original. I wonder how much compute would be required to make the offset far enough to hold up from scrutiny, and in court.
Alternatively, you could manually change the network model, add a few hidden layers, etc... modifying the parameters in step, and result in a new model and new parameters. Some training to vary the parameters, and it's now a new work.
I would hazard a guess here that taking the parameters and continuing training constitutes "using" the parameters. Then when you get the subpoena you would have to explain the thousands of emails and slack messages discussing how you extend their parameters... :)
It's counterintuitive for me that this is a problem. One man should be able to do this in a month, no need for coordination that leaves behind evidence. Besides, how hard is it to communicate in secret?
I am a structural biologist. This is one of the handful of topics that overlaps with my field here. I'm very excited to play with this, although it might eventually put me out of a job.
Here's where I think we need to be going: You go to a doctor's office, sick. 1) They take a blood sample. 2) They find the malignant bacteria and DNA sequence it. 3) If it's a known strain, they know what antibiotics to use on it. 4) If not, they solve protein folding on the genes. 5) From that, they see which existing antibiotics would kill it. 6) If none will, then given the proteins, they have to derive a new antibiotic.
1) is easy. 2) might not be - there can be a lot of things in a blood sample, and finding only the interesting (bad) things might not be simple. The sequencing part is pretty much solved. 3) would take a bit of work, but I think it's possible now. 4) we're getting there. 5) might have a fair amount in common with 3), but it probably takes some additional work. 6) is... probably non-trivial.
That's just one research agenda. There are others. You may have to move to related work, but I doubt you're going to be out of a job in this lifetime.
Basically sequence everything what's in your blood and look for what doesn't match your genome === infection. The problem is this is orders of magnitude more compute intense than whole genome sequencing. Basically increased demand for sequencing far outmatches available compute!
DNA sequencing is still slow and very expensive. On the scales you're talking about it's just not worth it.
I think I agree at a high-level that there is a huge reservoir of demand for this technology. But it's also possible that solving protein folding and similar research will simply cease to be a bottleneck for that demand, and people will be out of jobs.
The implicit assumption you are making is that the demand increases in lock step with productivity gains.
100x faster drug discovery, 100x more drugs need to be discovered => same number of people employed.
These correlations do hold for technical fields, but logically there should be a point beyond which productivity gains outpace, demand growth / demand could even stop growing. One should either retool to solve a newer problem before this point is reached, or hope that the point is not reached in the span of their career.
Oil rig builders for example - manufacturing has been increasingly automated, but the demand for oil rig building has grown consistently. But they should probably look into solving other problems given that demand is shifting.
>but logically there should be a point beyond which productivity gains outpace
The limiting factor on drug approval is clinical trials. Once every living person is enrolled in a clinical trial, we will have hit the maximum rate at which humanity can produce new drugs.
That might be more than 10x the current rate, but probably less than 1000x.
The number of people needed for a trial depends on the size of the effect.
I think we have a warped idea of that, because of the practice of looking for a barely "statistically significant" effect from a substance that isn't really understood.
If you have something that just eradicates a disease immediately, because you really understand what you are doing, you don't need very many tests to confirm it works.
In principle you could put people into multiple trials and gain somewhat additional throughput. Google implemented putting users into multiple different experiments (paper by Tang et al) and that made a huge difference.
It would both become a tool he used (to produce initial structures to fit in density maps) and a tool that used his or her output (because alphafold requires known protein structures that are homologous to the one you're predicting).
Right now I make my living cloning, expressing, and purifying proteins, crystallizing them or freezing them onto EM grids, and solving their structure. From start to finish, it's months to years of work for each structure.
Could you give an overview of how people can leverage this (or how you might?).
From reading around about it, it sounds like there's often a need to find a certain type of molecule to activate/inhibit another based on shape and the ability to programmatically solve for this makes the searching way easier.
Is this too oversimplified/wrong? How will this be used in practice.
I've worked in bio and drug discovery for some 25 years. That includes building classifiers using gradient descent in the 90s (when algorithms, computers and data were all much worse). I ported DOCK to Linux in ~96 or 97. Since then I built an academic and then industrial career with some emphasis on using computing to solve problems in drug discovery, but I don't play that role any more.
It doesn't look like the models produced by this would immediately turn the challenging problem of finding, approving, and marketing successful pharmaceuticals (IE, it doesn't eliminate any real bottleneck).
There was a long-term dream of structure-based drug discovery based on docking, but IMO, it has never really proved itself (most of the examples of success are cherry picked from a much larger pile of massive failures).
Because programming computers is far more lucrative, and I'm better at it. However, if I had an unlimited budget I would return to biology.
I spent 15 years trying to be a professor and failed miserably. I was bad at it and didn't like what professors have to do.
I then moved to industry to be a random engineer and thrived doing things entirely unrelated to drug discovery. Eventually, I convinced my company to invest heavily in life sciences. This was successful and I was on track to be a powerful player (a "research engineer", just like the DM folks who are building these things) in this space, when the project got very popular and I was elbowed aside by others who are more aggressive. So I went back to being a programmer again, it's much less stressful, pays better, and realistically, much of my time is just telling scientists what I would do if I was in their place anyway.
"Don't swim with the sharks if you don't like being bitten"
> much of my time is just telling scientists what I would do if I was in their place anyway.
That sounds familiar. I guess they mostly don't listen, whatever your record -- especially if it was in a different field they could learn from -- but I hope it's not always like that.
Maybe a culture clash? Academia is all about status and prestige - more often scientific outcomes seem to be a means to get the former (why journals don't publish negative results, why studies fail to replicate, why stuff isn't open access, why people worry about getting scooped, etc.)
Tech (at its best) hates credentialism (sometimes I think to a point of over-correction).
That said, 80% of the devs in the bay area seem to have gone to Stanford or MIT, so...
I haven't worked on the drug-side of things, but here my bio perspective: It's kind of out-of-vogue, but consider the "lock and key" model of proteins and small molecules (drugs). For drug design, what you want to do is get a key that fits just one lock (to pull whatever lever) and not others (to avoid side-effects). It's relatively easy to find a molecule that fits a protein, because that protein is what you might spend years researching and probing, but it's tricky to check if it does anything against ~100,000 others in humans. If you could do an in silico computational survey to be like, oh, maybe it'll target this accidentally, you could spot-check those in vitro, and/or stick on some other atoms to your small-molecule to make it not fit that off-target.
Holy grail, IMO, though is being able to design de novo protein sequences (to make "biologics", aka engineered protein drugs) that can a) target (bind/block/enhance) or do (chemical reactions) what you want and only that, b) are easily synthesizeable by bacteria/yeast (cheap to make), and c) are stable (easy to transport/store).
> Could you give an overview of how people can leverage this (or how you might?).
Short answer: nobody knows. Traditionally, protein folding is a solution in search of a problem, but that's largely because the predictions were...unusably bad. This was always more of a super-difficult validation problem for the force fields and simulation methods, which could then be used for other problems of greater value (such as rational protein design, or simulation of the motion of proteins with known structures).
These predictions are better, but still pretty far from the level of precision that you'd want for any kind of rational drug design, where the exact locations of protein side-chains (for example) matter a lot. You'll note that AlphaFold returns structures that are "relaxed" using one of the oldest simulation systems for proteins: AMBER. So it's not exactly a clean-room solution to the problem, and you can't assume that the details (which matter to drug design) are going to be any better than for the older methods.
But that said, if you have a method that can reliably give you a blurry view of the overall shape of a protein, even that could be useful for things like target discovery or inference of biological networks. But this is still a lot closer to pure research than "revolutionizing drug discovery", as is frequently batted around on reddit, HN and the press.
Also I would say that really they just made improvements to protein structure prediction, not protein folding which is the dynamic process by which proteins reach their equilibrium fold.
I do agree with this but would add that you wouldn't want to do static docking to a single protein and sidechain configuration anyway so you're bound to find a usable forcefield. The incoming structure (ideally!) only needs to fall into the right energy landscape valley. If your forcefield and MD simulation can't keep this stable within the natural protein's configuration space you are probably not going to make progress, and you need the simulations to evaluate the natural energy landscape at body temperature, not cryo temperature.
There are some examples of this issue in the AlphaFold blog, some protein loops that they thought were mispredicted but it turned out they were part of an energy degeneracy so the natural state fluctuated pretty wildly, so if you can't simulate this properly it matters less how accurate the incoming structure is (to a certain degree of course).
Any drug target of any real-world interest is going to have local motion (i.e. floppiness that is dependent on circumstance and time) that matters at least as much to the finding of a drug than the fold of the protein itself.
One place I hesitate here is that step 0 to doing that kind of a simulation is having a decent starting structure. So maybe protein folding can help there. But I'm also skeptical, because the quality of the structure matters a great deal to getting a good simulation. A bad/blurry structure is of marginal use.
You can't do intelligent drug design if you don't know what the target protein looks like. We've gotten great at solving protein structures with things like crystallography and cryo-EM microscopy. Unfortunately, many interesting drug targets reside in the membrane of a cell, which means you can't easily work with them in a lab because they aren't soluble in anything but a plasma membrane. For instance, this is an issue with the 5HT2A protein, a g coupled protein receptor that is implicated in many serotonin related pathways.
Being able to predict what it would look like would be a huge deal because then you can go about intelligently designing drugs for it.
It can be an aid in drug development, and can perhaps assist a bit in tuning small molecule drugs for more stable binding.
Though I think the major impacts will be two-fold:
(1) The field of structural biology is going to see a change, with much more data available. Some structures of difficult to crystallize proteins will be solved, which may lead to much greater biological understanding. We may enter a time, where once you have a primary sequence, you also have a likely 3d-structure, which will probably change the daily work of quite a few biologists a bit.
(2) Industrial protein design. A tool such as this can potentially have great utility in optimizing proteins as chemical catalysts for various processes in different industries. This includes expanding the conditions under which a protein is active and also making their conformation more stable and so the protein more long-lived in solution.
For those that are unaware, industrial protein design is a multibillion dollar industry. For example, decades ago Genentech and Dow Corning formed a company that developed proteases (proteins that cut other proteins) that worked at much higher temperatures than the ones in nature. This was then sold to P&G and other major laundry companies (laundry detergent contains idle enzymes activated by the heat of the laundry water, and they go clean up. "Protein gets out protein" was the marketing jingle.
That was a few billion dollars right there and almost all the work was done by hand by lab scientists.
I work in cancer research with a drug discovery focus in a lab with some structure biologists. My understanding is that if we identified proteins targets suitable for therapeutics then understand its structure to identify secondary binding sites could be crucial for drug discovery. Drugs can then be designed to modulate its biological functions.
Honest question: since AlphaFold doesn't really _solve_ the protein folding problem (it's NP-complete after all), but only _approximates_ solutions very well, what are the real impacts of this? Isn't a good approximation of a protein enough to cause unexpected problems? How do we know that an approximate structure will perform the same as the correct solution?
Protein folding is a physical/biological phenomenon. AFAIK we don't currently have a proper exact mathematical formulation of the problem that would let one determine its complexity.
You may be referring to this paper [1]. It only claims that one particular optimization problem, believed to give a solution to protein folding problems, is NP-hard. So, even if a suitable exact formulation exists, it is not yet proven that protein folding is hard, although it for sure seems plausible.
By the way, it is perfectly possible today to solve some very large-scale NP-hard problems (think millions of variables and constraints) in reasonable amounts of time (think minutes or hours). Examples are knapsack problems, SAT problems [2], the Traveling Salesman Problem [3] or more generally Mixed Integer Programming [4].
[1] "Complexity of protein folding", 1993, by Aviezri S. Fraenkel
The protein folding problem is not NP complete. The "formal" protein folding problem, as posed (find the set of dihedral angles whose resulting structure has the lowest energy) might be, but that bears only a distant resemblance to how people "solve" the problem today. At the very least, the statement is incorrect because many proteins don't actually fold to their energy minimum, they get stuck in kinetic traps, and the formal PF defintion never accomodated that idea.
Not really an answer to your question, but is the problem really NP-complete, or just combinatorially difficult? For example how is this condition of NP-completeness satisfied?
> it is a problem for which the correctness of each solution can be verified quickly [0]
I dont know much about protein folding, but for most things in life,exact solutions to NPC problems usually aren't needed for non-contrived problems. In many cases, approximations are good enough.
Besides, this is real life - if predictions and real life match, that's great. If they don't, well you know you went wrong somewhere.
I would upvote this twice if I could. Life science quite often NP-hard still approximate results are extremely useful.
Joke, which I think is from Sean Eddy (hammer).
Bioinformatics approaches a Computer Scientist for help with a hard problem. CS agrees to help. Year later CS comes back very excitedly. "your problem is not hard it is NP-hard!". Bioinformatics nods, and says "I still got to solve it" and continues finding ever faster and better approximations ;)
Also problem space is both bounded (you don't have infinite length proteins) and f'd up in reality. e.g protein hijacking and re-conformation in the face of an infectious agent.
A very-non-expert opinion, if an approach approximates it pretty well and can be improved upon, then it could end up being quite useful. Given that biology exists on a real, tangible scale then perfection in the fold prediction isn't necessary, instead just an approximation that is sufficiently good to be functionally useful.
^ That sounds like word-salad BS but I think there's some truth to it. I know protein folding has been postulated to be useful in terms of understanding basic biology, understanding disease pathology, and drug prediction. While a wide range of approximations are functionally useless, perhaps the Alphafold approach or some improved version of it surpasses the functionally useful threshold.
Yes, it is still useful. Even structures obtained through traditional means (eg. x-ray crystallography) are approximations to an extent since there are limits to the resolution that you can obtain and oftentimes regions of proteins are "disordered". Additionally, these structures are only snapshots of a protein in a particular state, which may not completely reflect the dynamics of the protein in its native environment.
Kind of disagree with this.. solving protein structures is not the rate limiting step in drug discovery or in biochemical assays -- not by a long shot. See this excellent comment by @dekhn on a related submission: https://news.ycombinator.com/item?id=27849046
I would expect that once AlphaFold has helped you identify a potential protein (e.g. as a drug) out of a bigger set of potential proteins, there will still be a manual step of traditional cryoEM, NMR, etc. to get an accurate high-resolution structure.
To me, the interesting thing is not the specific results but rather that you can accurately predict crystal structures from sequence alone. This begets the question: what other physical biological properties can we predict?
AlphaFold is not about solving any kind of NP-complete problem.
Proteins consist of chains of amino acids which spontaneously fold up to form a structure. Understanding how the amino acid chain determines the protein structure is highly challenging, and this is called the "protein folding problem".
People use mathematical models to predict how proteins fold in nature. Many such mathematical models are stated in terms such as "proteins fold into a configuration that minimizes a certain energy function". Even the simplest such models [1] give rise to NP-hard decision problems, which are also known (somewhat confusingly) as "protein folding problems". To make this a bit less confusing, I will call the mathematical decision problems PFPs.
Like all mathematical models, our protein folding models don't correspond exactly to reality. Even if you are somehow able to determine the exact mathematical solution to a mathematical PFP, that _still_ doesn't guarantee that the real protein that you were trying to model behaves like the mathematical solution would indicate. E.g. the protein may fold in such a way that it gets stuck in a local optimum of the energy function you were using.
How do we detect this? We make inferences about how the protein should behave, given the mathematical solution to the Protein Folding Problem, and then we perform experiments, and find out (empirically) that the protein behaves in a manner that is inconsistent with the inferences drawn from the mathematical model. Scientists _do_ do this. And they would have to do it even if they had a fast, exact way to solve NP-complete problems, because the NP-complete problems are still just part of a mathematical model, and need not correspond to reality in any way.
The success of AlphaFold is not measured by how well it solves (or approximates) mathematical PFPs. The success of AlphaFold is measured by making successful predictions about how certain proteins will fold. And this is exactly how it was tested [2]: they threw it at a bunch of problems for which scientists have empirically determined how certain amino acid chains fold, but didn't release the results. And then they compared the solutions predicted by AlphaFold, and found that most of the predictions were consistent with what they knew to be the case.*
The model parameters are only available for non-commercial use. That's a shame, as I presume there might be a lot of medical startups that would benefit from having this kind protein-folding tech available.
Unless I'm mistaken, you could train the model yourself, starting with a random set of values. In time, your error rates would be low enough to have a new set of parameters which you could use however you like.
Yep, but there's a couple of problems. Firstly, AFAIK Deepmind haven't made all the code and settings they used to train the model available (although the paper does describe the architecture). Secondly, training a machine-learning model of this complexity is generally much more expensive, in terms of time and compute requirements, than using the resulting model.
If you're a a medical startup, having an off-the-shelf prediction model you can just start using for all your protein folding needs is a very different proposition from having to train one yourself from scratch.
That said, hopefully other researchers and institutions will take Google's research and produce an equivalently powerful model but with a more commercially-friendly open-source license. From some comments in this thread, it sounds like that's already happening, in fact.
> The simplest way to run AlphaFold is using the provided Docker script. This was tested on Google Cloud with a machine using the nvidia-gpu-cloud-image with 12 vCPUs, 85 GB of RAM, a 100 GB boot disk, the databases on an additional 3 TB disk, and an A100 GPU.
Working with one of the team providing the uniref dataset used here. Running any kind of torrent stuff in an university network setting is a central policy fight one just does not want to get into at all.
On the other hand outbound network traffic from an university is "free". So the benefit is absolutely minimal from a hosting perspective.
Where there isn't an available crystal structure, Alphafold can be used to create initial structures for simulation via folding@home, replacing older homology modeling techniques.
no, it wouldn't make sense to do that. Folding@Home is for ab initio where you don't have any prior info for the structure, this is for homology modelling. F@H probes the dynamics of protein folding, this just makes a static prediction.
Honest question: since AlphaFold doesn't really _solve_ the protein folding problem (it's NP-complete after all), but only _approximates_ solutions very well, what are the real impacts of this? Isn't a good approximation of a protein enough to cause unexpected problems? How do we know that an approximate structure will perform the same as the correct solution?
There is a lot of bias in the chat here from a more chemistry and pharma slant. If you ignore this AlphaFold solves in a very meaningful way the problem blocking a lot of science investigation.
For comparative and evolutionary analysis structure is far more conserved than sequence. Especially in things like viruses or anything with a high rate of reproduction like bacteria. Just knowing the general fold or overall structure is enough to do structural alignment and tell if two genes are related on that basis, even if their genomic sequence is completely dissimilar. Large groups of researchers rely on sequence homology built from sequences of known structure.
But AlphaFold works well in new sequence space to far more accuracy than is needed. If we had an AlphaFold prediction for every known sequence suddenly the evolutionary relationships between all genes and even all species would be far clearer. This on its own unlocks a new foundation to reason about function and molecular interaction with a wholistic systems view without gaps in what we can know with some reasonable assurance.
For an analogy think of the difference between having books in different languages describing objects. You know what some of the book in English might say but you dont even know if the book in Spanish is even talking about the same things. AlphaFold is like an AI that transforms all the books into picture books and now we can use image similarity or have one person look at all pictures.
{kind=link}
https://www.nature.com/articles/s41586-021-03819-2_reference...